
Research
The Kwapis Lab aims to answer one big-picture question: How does long-term memory work at a molecular level in the young and old brain? We want to understand the epigenetic and transcriptional mechanisms that underlie memory formation, storage, and updating and identify how these mechanisms change in old age and in various disease states.

Our research uses cutting-edge techniques to dissect the molecular mechanisms that underlie memory and age-related memory decline. These techniques include:
-in vivo CRISPRi and CRISPRa to reduce or drive transcription in a site- and circuit-specific manner
-Cloning, cell culture, and viral vectors to manipulate gene expression or activity
-CatFISH and viral TetTag to capture and visualize active neuronal ensembles and chemogenetics to manipulate those ensembles
- Molecular and cellular biology techniques including RT-qPCR, western blots, immunofluorescence, chromatin immunoprecipitation, RNA-seq, etc.
-Sophisticated behavioral techniques, including fear conditioning, stress-enhanced fear learning (SEFL), object location/recognition memory, memory updating paradigms, etc.
Current Projects
Age-related epigenetic repression of Per1 could have different outcomes depending on the brain region affected. 1: Urban et al., 2021; Brunswick et al., 2023; 2: Kwapis et al., 2019; Bellfy et al., 2023
1. Understanding the mechanisms that exert diurnal control over memory, including how these processes differ by age and sex
Humans and other animals have internal circadian clocks that drive the rhythmic cycling of biological processes across the 24h day. These circadian rhythms influence a number of physiological events, including memory formation. Interestingly, in normal aging and in most major neuropsychiatric conditions, there are characteristic impairments in both memory and the circadian rhythm, suggesting an underlying connection.
We have discovered a novel mechanism that may serve as a key interface between the circadian system and long-term memory formation: the clock gene Period1 (Per1). Our work has shown that Per1 functions in the dorsal hippocampus to modulate spatial memory consolidation in addition to its well-documented role in the brain’s central pacemaker, the suprachiasmatic nucleus. Per1 is also repressed in the dorsal hippocampus and retrosplenial cortex in old mice that have spatial memory impairments and upregulating Per1 within either of these brain regions can ameliorate age-related impairments in memory . Together, this work suggests that Per1 ‘moonlights’ in memory-relevant brain regions to exert diurnal control over memory and age-related alterations in Per1 in these regions may contribute to age-related memory decline.
Our most recent work shows that female mice do not display diurnal oscillations in memory, an effect that may be explained by high levels of the memory-promoting sex hormone estradiol. Ongoing work in the lab is testing this hypothesis, as well as how Per1 and memory change in pre-pubescent adolescent mice.
2. Investigating how existing memories are updated with new information
One aspect of memory that is especially vulnerable to aging is the ability to update memories. Memories are not stored as fixed records of experience but are dynamically modified to incorporate new information. Memory updating is impaired in aging, yet very little is known about the mechanisms that go awry, in part because most laboratory paradigms focus on new memory formation. We have developed and validated a new memory updating task called Objects in Updated Locations (OUL) to specifically understand the mechanisms behind memory updating.
Using OUL, we have shown that the repressive histone deacetylase HDAC3 contributes to age-related impairments in memory updating, possibly by restricting the transcriptional program needed to modify memory or by promoting storage of the memory updating in the same neuronal ensemble storing the original memory. We are currently testing whether improving neuronal co-allocation (i.e. encouraging the update to be stored in the original-memory ensemble) ameliorates age-related impairments in updating. We are also testing whether we can leverage memory updating to produce more permanent extinction of fear memories.
Viral TetTag combined with cFos labelling to identify neurons engaged by initial memory formation and subsequent updating.
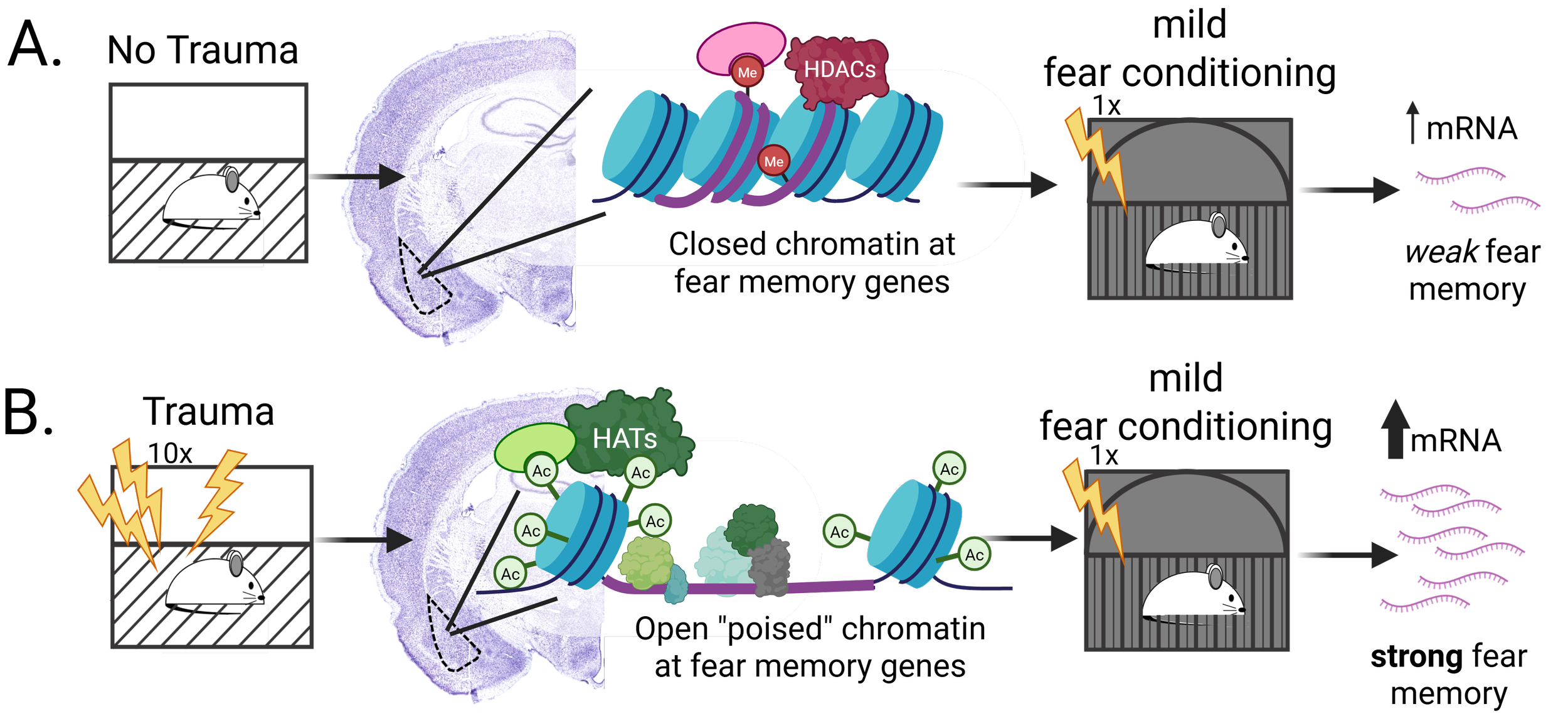
Proposed mechanism underlying the SEFL paradigm. A) In the absence of the trauma event, HDAC3 maintains a restrictive chromatin state in the basolateral amygdala, minimizing mRNA expression during subsequent fear conditioning and forming a very weak fear memory. B) Following the trauma event, HDAC3 is kicked off of the chromatin in the basolateral amygdala, permitting extensive acetylation and an open chromatin state that allows for substantial mRNA expression during subsequent fear conditioning and the formation of a strong fear memory.
3. Identifying sex-specific mechanisms driving stress-enhanced fear learning
Fear is critical to survival, but excessive fear is problematic when it interferes with normal functioning. Exaggerated fear is a hallmark of post-traumatic stress disorder (PTSD) in which an emotionally traumatic event predisposes an individual to show excessive responding to subsequent mild stressors. Roughly 7% of people in the US will experience PTSD at some point, with women nearly twice as likely as men to develop this disorder, but little is known about the molecular underpinnings and why females are more susceptible.
We are actively investigating whether trauma establishes a set of epigenetic marks at key genes important for fear memory, priming these genes so that subsequent stress can more efficiently drive their expression. Using the stress-enhanced fear learning (SEFL) paradigm, we are testing the molecular and epigenetic mechanisms that support SEFL and investigating how these mechanisms are altered in females to promote fear sensitization.